C. Liu, W. Huhn, K. Du, A. Vazquez-Mayagoitia, D. Dirkes, W. You, Y. Kanai, D. Mitzi, and V. Blum, Tunable Semiconductors: Control over Carrier States and Excitations in Layered Hybrid Organic-Inorganic Perovskites, Physical Review Letters 121, 146401-1‑146401-6 (2018). doi: 10.1103/PhysRevLett.121.146401.
Methylammonium lead iodide: atomic structure
See all entries for this property (35 total)
Origin: computational
Crystal system: orthorhombic
| a: | 9.25 Å |
| b: | 12.88 Å |
| c: | 8.62 Å |
| α: | 90° |
| β: | 90° |
| γ: | 90° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: PBE
Level of relativity: atomic ZORA
Basis set definition: tight
Numerical accuracy: tight
Comment: local structure optimization: unit cell + atomic positions [unconstrained] Files available on NOMAD: http://nomad-repository.eu:8080/NomadRepository-1.1/doi/result/index.zul?dataset=5842555
Entry added on: March 15, 2019, 12:04 a.m.
Entry added by: Xiaochen Du Duke University
Last updated on: June 22, 2022, 10:03 p.m.
Last updated by: Rayan C Duke University
Download data
Methylammonium lead iodide: atomic structure
See all entries for this property (35 total)
Origin: computational
Crystal system: orthorhombic
| a: | 8.99 Å |
| b: | 12.71 Å |
| c: | 8.48 Å |
| α: | 90° |
| β: | 90° |
| γ: | 90° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: PBE with Tkatchenko-Scheffler van der Waals correction
Level of relativity: atomic ZORA
Basis set definition: tight
Numerical accuracy: tight
Comment: local structure optimization: unit cell + atomic positions [unconstrained] Files available on NOMAD: http://nomad-repository.eu:8080/NomadRepository-1.1/doi/result/index.zul?dataset=5842555
Entry added on: March 15, 2019, 12:04 a.m.
Entry added by: Xiaochen Du Duke University
Last updated on: Sept. 7, 2021, 5:51 p.m.
Last updated by: Rebecca Lau Duke University
Download data
Methylammonium lead iodide: atomic structure
See all entries for this property (35 total)
Origin: computational
Crystal system: orthorhombic
| a: | 9 Å |
| b: | 12.72 Å |
| c: | 8.48 Å |
| α: | 90° |
| β: | 90° |
| γ: | 90° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: PBE with many-body dispersion (range-separated MBD@rsSCS)
Level of relativity: atomic ZORA
Basis set definition: tight
Numerical accuracy: tight
Comment: local structure optimization: unit cell + atomic positions [unconstrained] Files available on NOMAD: http://nomad-repository.eu:8080/NomadRepository-1.1/doi/result/index.zul?dataset=5842555
Entry added on: March 15, 2019, 12:04 a.m.
Entry added by: Xiaochen Du Duke University
Last updated on: Sept. 7, 2021, 5:51 p.m.
Last updated by: Rebecca Lau Duke University
Download data
5,5‘‘‘-bis(aminoethyl)-2,2‘:5‘,2‘‘:5‘‘,2‘‘‘-quaterthiophene lead bromide: atomic structure Verified
See all entries for this property (4 total)
Origin: computational
Crystal system: triclinic
| a: | 39.95010996 Å |
| b: | 11.60299493 Å |
| c: | 11.48001227 Å |
| α: | 89.98225306° |
| β: | 91.24294626° |
| γ: | 90.02085902° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 11, 2019, 11:36 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-thiophene lead bromide: atomic structure Verified
all-anti-(AE1T)PbBr4
Origin: computational
Crystal system: triclinic
| a: | 26.00142723 Å |
| b: | 11.8676782 Å |
| c: | 11.20758355 Å |
| α: | 90.00437713° |
| β: | 89.1487017° |
| γ: | 90.00031215° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the AE4T organic part by all-anti AEnT molecules. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: April 9, 2022, 1:03 p.m.
Last updated by: Rayan C Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-bithiophene lead bromide: atomic structure Verified
all-anti-(AE2T)PbBr4
Origin: computational
Crystal system: triclinic
| a: | 30.19597054 Å |
| b: | 11.80477997 Å |
| c: | 11.35930053 Å |
| α: | 90.0693883° |
| β: | 89.44587558° |
| γ: | 90.17116074° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the AE4T organic part by all-anti AEnT molecules. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 11, 2019, 11:37 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-terthiophene lead bromide: atomic structure Verified
all-anti-(AE3T)PbBr4
Origin: computational
Crystal system: triclinic
| a: | 35.51857397 Å |
| b: | 11.87626018 Å |
| c: | 11.47215253 Å |
| α: | 90.42074456° |
| β: | 91.29657434° |
| γ: | 87.62408207° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the AE4T organic part by all-anti AEnT molecules. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 11, 2019, 11:37 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5‘‘‘-bis(aminoethyl)-2,2‘:5‘,2‘‘:5‘‘,2‘‘‘-quaterthiophene lead chloride: atomic structure Verified
See all entries for this property (2 total)
Origin: computational
Crystal system: triclinic
| a: | 40.85056467 Å |
| b: | 11.29489724 Å |
| c: | 10.94887555 Å |
| α: | 90.01889107° |
| β: | 91.755904° |
| γ: | 89.98766217° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the Br atoms with Cl. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 11, 2019, 11:12 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5'''-bis(aminoethyl)-2,2':5',2'':5'',2'''-quaterthiophene lead iodide: atomic structure Verified
See all entries for this property (3 total)
Origin: computational
Crystal system: triclinic
| a: | 39.01481456 Å |
| b: | 12.09647627 Å |
| c: | 12.22481263 Å |
| α: | 90.0346079° |
| β: | 91.076978° |
| γ: | 90.03861881° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the Br atoms with I. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: Aug. 11, 2019, 4:54 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-quinquethiophene lead bromide: atomic structure Verified
all-anti-(AE5T)PbBr4
Origin: computational
Crystal system: triclinic
| a: | 45.4903936 Å |
| b: | 11.95970872 Å |
| c: | 11.37132315 Å |
| α: | 88.4690582° |
| β: | 89.29838954° |
| γ: | 88.7641499° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: with spin-orbit coupling
Comment: Using the experimental structure of AE4TPbBr4 [1], also in Dataset ID 217. Replace the AE4T organic part by all-anti AEnT molecules. Refer to SI Part IX for more details. [1] D. B. Mitzi, K. Chondroudis, and C. R. Kagan, Inorg. Chem. 38, 6246 (1999).
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 11, 2019, 11:17 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5‘‘‘-bis(aminoethyl)-2,2‘:5‘,2‘‘:5‘‘,2‘‘‘-quaterthiophene lead bromide: band structure Verified
See all entries for this property (3 total)
Experimental then geometry optimized
Origin: computational
Crystal system:
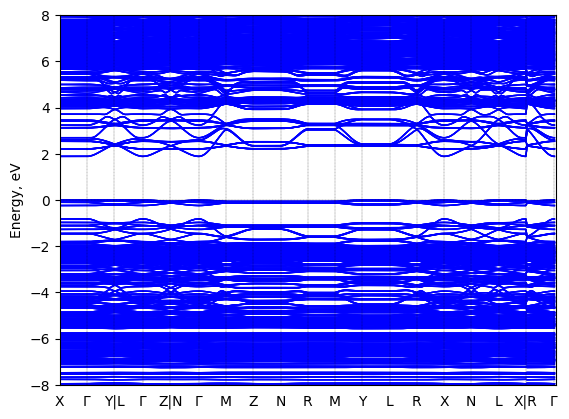
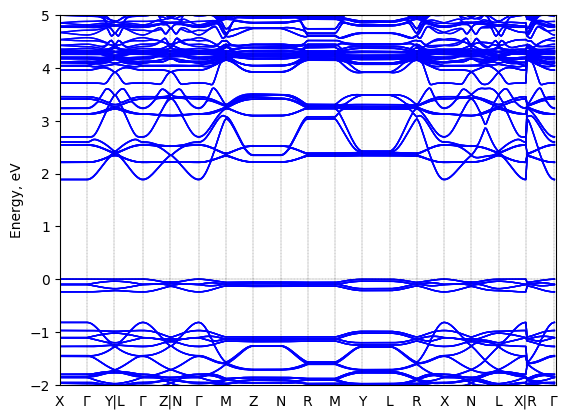
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:01 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: March 23, 2022, 5:59 p.m.
Last updated by: Rayan C Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5‘‘‘-bis(aminoethyl)-2,2‘:5‘,2‘‘:5‘‘,2‘‘‘-quaterthiophene lead bromide: band structure Verified
See all entries for this property (3 total)
Theory then geometry optimized; all-anti-AE4TPbBr4
Origin: computational
Crystal system:
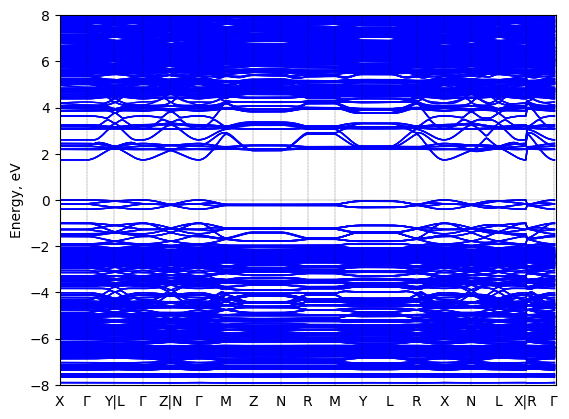
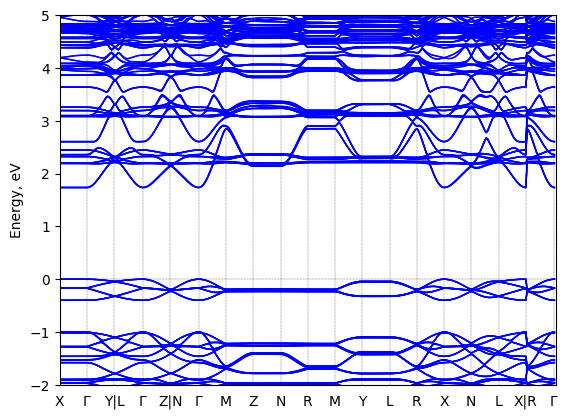
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:02 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: March 23, 2022, 6 p.m.
Last updated by: Rayan C Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-thiophene lead bromide: band structure Verified
Theory then geometry optimized
Origin: computational
Crystal system:
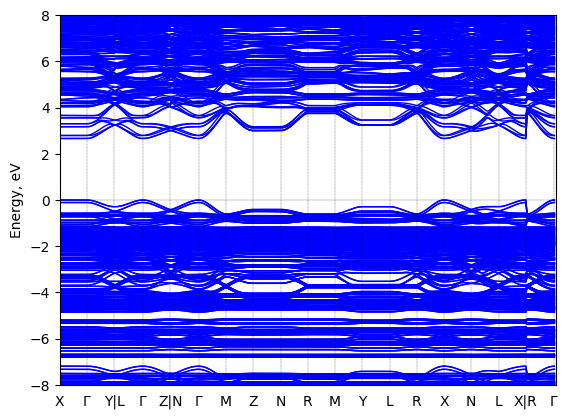
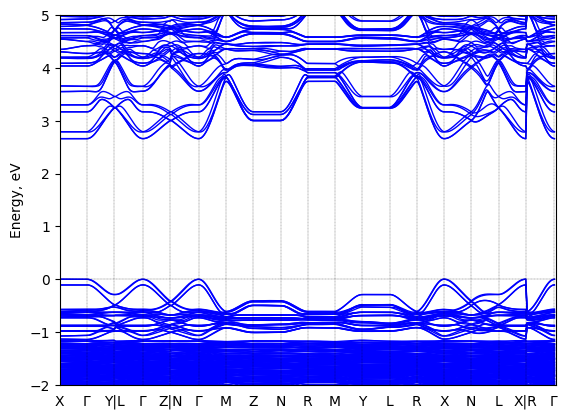
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 1x2x2
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:03 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: Oct. 3, 2022, 10:36 p.m.
Last updated by: Harrison York Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-bithiophene lead bromide: band structure Verified
Theory then geometry optimized
Origin: computational
Crystal system:
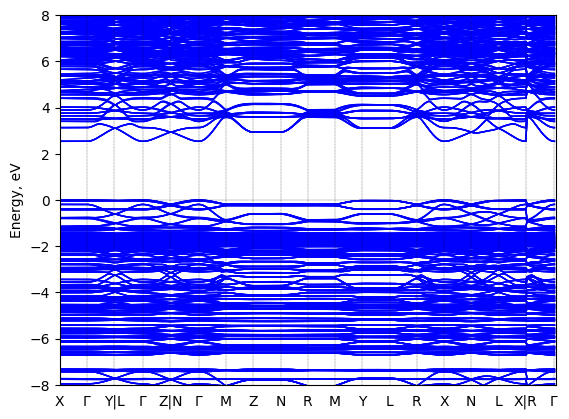
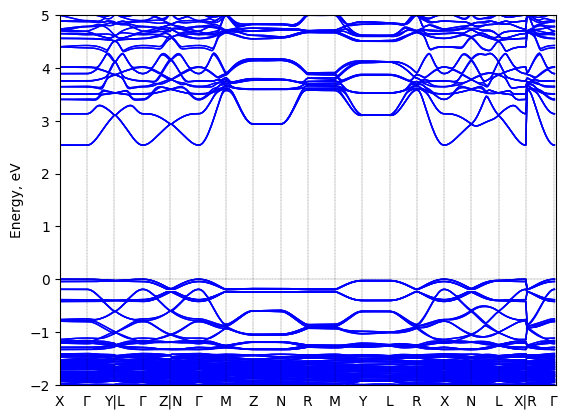
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:03 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: May 8, 2019, 4:03 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-terthiophene lead bromide: band structure Verified
Theory then geometry optimized
Origin: computational
Crystal system:
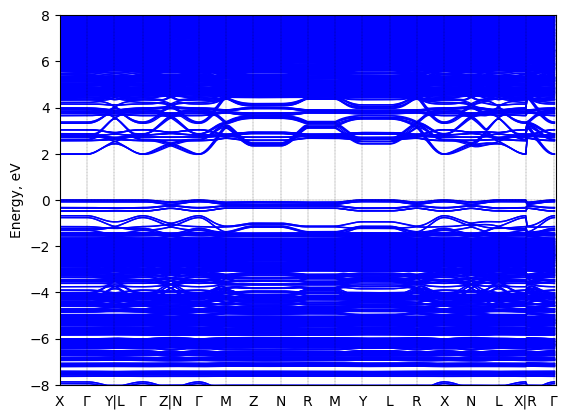
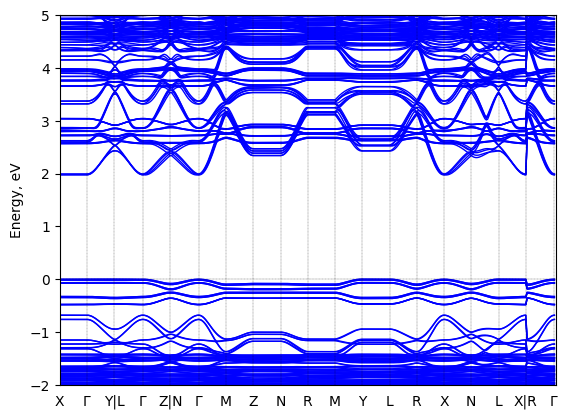
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:03 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: May 8, 2019, 4:03 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5‘‘‘-bis(aminoethyl)-2,2‘:5‘,2‘‘:5‘‘,2‘‘‘-quaterthiophene lead chloride: band structure Verified
See all entries for this property (2 total)
Theory then geometry optimized
Origin: computational
Crystal system:
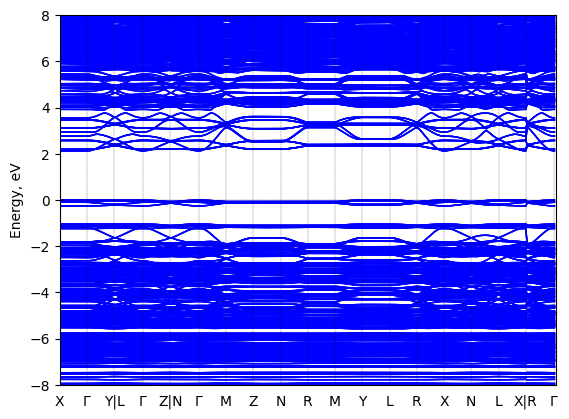
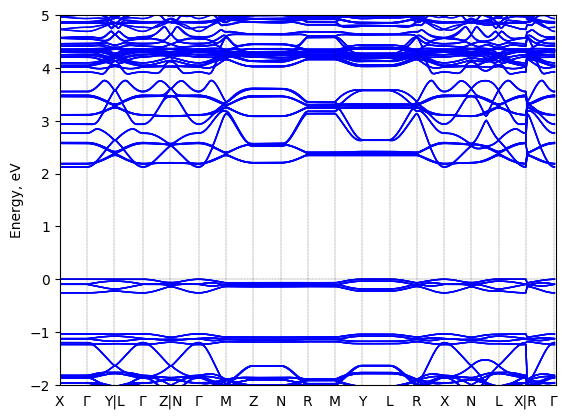
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:04 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: May 8, 2019, 4:04 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Bis(aminoethyl)-quinquethiophene lead bromide: band structure Verified
Theory then geometry optimized
Origin: computational
Crystal system:
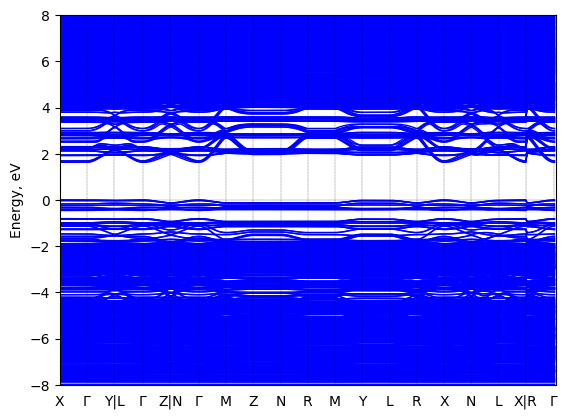
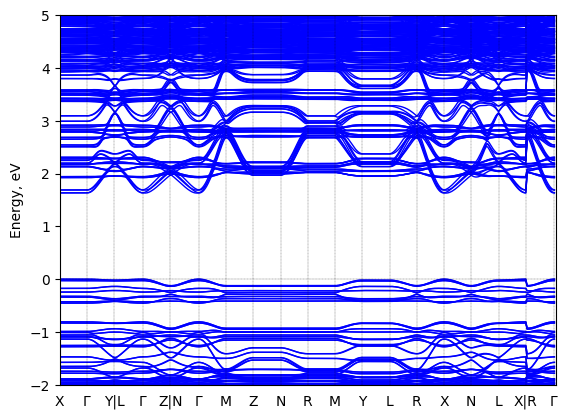
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: May 8, 2019, 4:04 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: May 8, 2019, 4:04 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Methylammonium lead iodide: band gap (fundamental)
See all entries for this property (6 total)
Fundamental band gap
Origin: computational
Crystal system: orthorhombic
| Band gap (fundamental), eV |
|---|
Crystal system: orthorhombic
| Band gap (fundamental), eV |
|---|
Crystal system: orthorhombic
| Band gap (fundamental), eV |
|---|
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
Level of relativity: atomic ZORA with spin-orbit-coupling
Basis set definition: tight
Comment: Files available on NOMAD: http://nomad-repository.eu:8080/NomadRepository-1.1/doi/result/index.zul?dataset=5842555
Entry added by: Xiaochen Du Duke University
Last updated on: May 27, 2019, 11:08 p.m.
Last updated by: Xiaochen Du Duke University
Download data
Methylammonium lead iodide: atomic structure
See all entries for this property (35 total)
MAPI relaxed geometry with PBE+TS
Origin: computational
Crystal system: orthorhombic
| a: | 8.986819693 Å |
| b: | 12.7083081 Å |
| c: | 8.476236171 Å |
| α: | 90.06532836° |
| β: | 90.02979487° |
| γ: | 90.04474512° |
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: PBE with Tkatchenko-Scheffler van der Waals correction
K-point grid: 1x2x2
Level of relativity: atomic ZORA
Basis set definition: tight
Numerical accuracy: tight
Comment: local structure optimization: unit cell + atomic positions [unconstrained]Files available on NOMAD: http://nomad-repository.eu:8080/NomadRepository-1.1/doi/result/index.zul?dataset=5842555
Entry added on: Feb. 21, 2020, 11:27 a.m.
Entry added by: Xixi Qin Duke University
Last updated on: May 14, 2021, 5:54 p.m.
Last updated by: Xixi Qin Duke University
Download data
See all entries for this property (3 total)
TEST
Origin: computational
Crystal system: triclinic
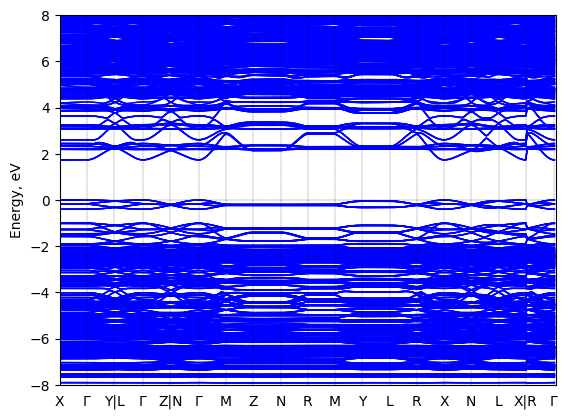
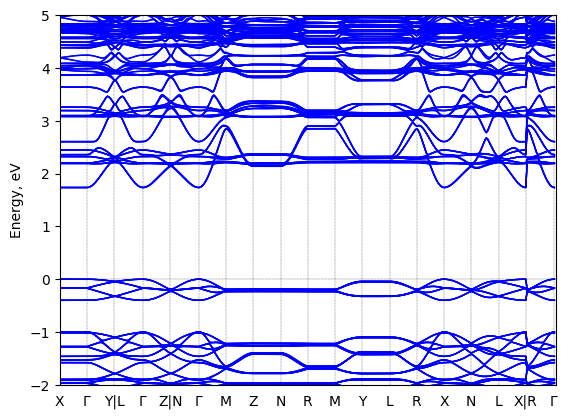
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: June 23, 2020, 7:38 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: March 23, 2022, 6 p.m.
Last updated by: Rayan C Duke University
Download data
5,5'''-bis(aminoethyl)-2,2':5',2'':5'',2'''-quaterthiophene lead iodide: band structure Verified
See all entries for this property (2 total)
Experimental then geometry optimized
Origin: computational
Crystal system: unknown
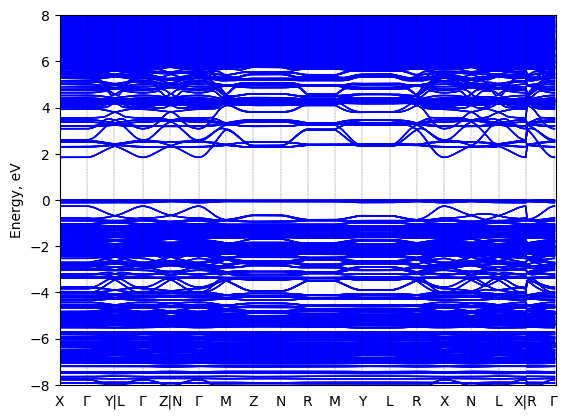
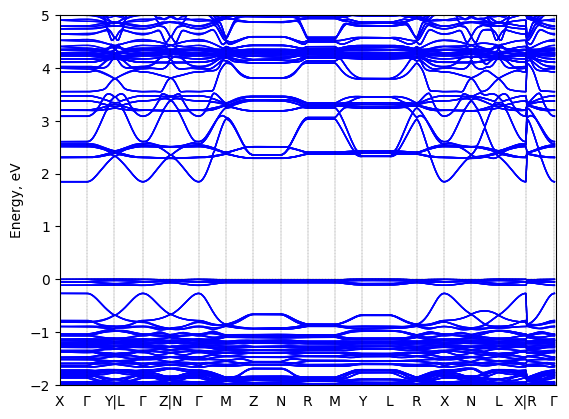
Sample type: single crystal
Code: FHI-aims
Level of theory: density functional theory
Exchange-correlation functional: HSE06 α = 0.25, ω = 0.11/bohr
K-point grid: 3x3x3
Level of relativity: atomic ZORA with spin-orbit coupling
Basis set definition: tight
Entry added on: July 21, 2020, 6:23 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: July 21, 2020, 6:23 p.m.
Last updated by: Xiaochen Du Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
5,5'''-bis(aminoethyl)-2,2':5',2'':5'',2'''-quaterthiophene lead iodide: atomic structure Verified
See all entries for this property (3 total)
Method: Single-crystal X-ray diffraction
Origin: experimental (T = 293.0 K)
Space group: C2/c
Crystal system: monoclinic
| a: | 38.779 (±0.003) Å |
| b: | 6.0863 (±0.0005) Å |
| c: | 12.3306 (±0.001) Å |
| α: | 90° |
| β: | 92.271 (±0.004)° |
| γ: | 90° |
- temperature = 293.0 K
Sample type: single crystal
Starting materials: PbI2 (99.999% trace metal basis), HI (57 wt. % in H2O, with hypophosphorous acid as stabilizer, assay 99.95%), N,N-Dimethylformamide (anhydrous, 99.8%, Sigma-Aldrich), 2-butanol (99.5%, VWR International)
Product: Orange crystals
Description: Synthesize AE4T·HI in the lab. References [1-3] Dissolve 2 mg PbI2 and 3 mg AE4T·HI in 0.7 ml DMF with a drop of HI. Then, layer 2 ml 2-butanol on top of the solution (SI Figure S8a). In the experiment, the target crystals came out after several days (Figure S8b).
Comment: References: [1] H. Muguruma, T. Saito, A. Hiratsuka, I. Karube, and S. Hotta, Langmuir 12, 5451 (1996). [2] H. Muguruma, T. Saito, S. Sasaki, S. Hotta, and I. Karube, J. Heterocyclic Chem. 33, 173 (1996). [3] H. Muguruma, K. Kobiro, and S. Hotta, Chem. Mater. 10, 1459 (1998).
Method: Single crystal X-ray diffraction
Description: Bruker D8 ADVANCE Series II at room temperature.
Comment: The unit cell parameters determined from this data are a = 38.779(3) °A, b = 6.0863(5) °A, c = 12.3306(10) °A, beta = 92.271(4) °, V = 2908.0(4) °A
Entry added on: Nov. 14, 2023, 1:50 p.m.
Entry added by: Volker Blum Duke University
Last updated on: Nov. 17, 2023, 5:33 p.m.
Last updated by: Rayan C Duke University
Data correctness verified by:
- Rayan C Duke University
Download data