These data sets are directly related (linked) to each other
Atomic structure
See all entries for this property (31 total)
Method: DFT-PBE plus Tkatchenko Scheffler dispersion, fully optimized
Origin: computational
Crystal system: triclinic
| a: | 32.35884857 Å |
| b: | 8.66248703 Å |
| c: | 8.698830605 Å |
| α: | 89.37708282° |
| β: | 85.6195755° |
| γ: | 85.42710876° |
Sample type: single crystal
- data set 225 (atomic structure)
- data set 1885 (atomic structure)
- data set 1901 (atomic structure)
- data set 1903 (atomic structure)
- data set 2267 (band gap (fundamental))
- data set 2269 (band structure)
Code: FHI-aims
Level of theory: DFT
Exchange-correlation functional: PBE+TS
K-point grid: 2*8*8
Level of relativity: atomic ZORA
Basis set definition: tight
Numerical accuracy: force convergence 5e-3 eV/AA
Comment: The original structure is based on the experimental resolved (PEA)2PbI4 structure from the reference tag, published in https://doi.org/10.1021/acs.inorgchem.7b01094
Entry added on: Feb. 25, 2020, 2:01 p.m.
Entry added by: Xixi Qin Duke University
Last updated on: March 6, 2023, 12:01 p.m.
Last updated by: Volker Blum Duke University
Download data
Atomic structure
See all entries for this property (31 total)
Origin: computational
Crystal system: triclinic
| a: | 32.36052322 Å |
| b: | 12.20835972 Å |
| c: | 12.34345722 Å |
| α: | 90.21797943° |
| β: | 83.75715637° |
| γ: | 89.88067627° |
Sample type: unknown
- data set 225 (atomic structure)
- data set 745 (atomic structure)
- data set 1901 (atomic structure)
- data set 1903 (atomic structure)
- data set 2267 (band gap (fundamental))
- data set 2269 (band structure)
Code: FHI-aims
Level of theory: DFT
Exchange-correlation functional: PBE (with TS scheme to account for the Van der Waals effect)
K-point grid: 2*4*4
Basis set definition: tight
Numerical accuracy: force convergence 5e-3 eV/AA
Comment: See Table S2 of Wright et al. (https://doi.org/10.1021/acs.chemmater.1c04213). This is the lowest-energy structure among multiple possible atomic structure models for (PEA)2PbI4 investigated in this reference. This structure was constructed based on the experimental c(2*2)*2 (PEA)2PbI4 published by Du. et al. (doi: 10.1021/acs.inorgchem.7b01094.)
Entry added on: Oct. 26, 2021, 1:38 p.m.
Entry added by: Xixi Qin Duke University
Last updated on: May 26, 2023, 2:53 p.m.
Last updated by: Volker Blum Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Atomic structure
See all entries for this property (31 total)
Origin: computational
Crystal system: triclinic
| a: | 12.34345739 Å |
| b: | 12.20835925 Å |
| c: | 32.36052428 Å |
| α: | 89.88067874° |
| β: | 83.75715796° |
| γ: | 90.21797821° |
Sample type: unknown
- data set 225 (atomic structure)
- data set 745 (atomic structure)
- data set 1885 (atomic structure)
- data set 1903 (atomic structure)
- data set 2267 (band gap (fundamental))
- data set 2269 (band structure)
Code: FHI-aims
Level of theory: DFT
Exchange-correlation functional: PBE
K-point grid: 4*4*2
Level of relativity: atomic ZORA with SOC
Basis set definition: tight
Numerical accuracy: force convergence 5e-3 eV/AA
Comment: This is a variant of structure 1885, rotated to use the c axis instead of the a axis as the out of plane axis. This structure is constructed based on the experimental c(2*2)*2 (PEA)2PbI4 published by Du. et al. (doi: 10.1021/acs.inorgchem.7b01094.)
Entry added on: Oct. 26, 2021, 4:37 p.m.
Entry added by: Xixi Qin Duke University
Last updated on: May 26, 2023, 3:13 p.m.
Last updated by: Volker Blum Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Atomic structure
See all entries for this property (13 total)
Origin: computational
Crystal system: triclinic
| a: | 11.72355567 Å |
| b: | 11.60903415 Å |
| c: | 32.86111038 Å |
| α: | 89.97165522° |
| β: | 84.12255457° |
| γ: | 90.31386573° |
Sample type: single crystal
- data set 225 (atomic structure)
- data set 745 (atomic structure)
- data set 1885 (atomic structure)
- data set 1901 (atomic structure)
- data set 2267 (band gap (fundamental))
- data set 2269 (band structure)
Code: FHI-aims
Level of theory: DFT
Exchange-correlation functional: PBE (with TS scheme to account for the Van der Waals effect)
K-point grid: 4*4*2
Basis set definition: tight
Numerical accuracy: force convergence 5e-3 eV/AA
Comment: This is a hypothetical, computationally generated structure that is intentionally NOT consistent with the experimental structure of (PEA)2PbBr4. Rather, the organic molecule configuration of this structure is borrowed from (PEA)2PbI4 structure (data ID 1901) but inorganic component is PbBr4, then fully optimized.
Entry added by: Xixi Qin Duke University
Last updated on: May 26, 2023, 3:26 p.m.
Last updated by: Volker Blum Duke University
Data correctness verified by:
- Rayan C Duke University
Download data
Band gap (fundamental)
See all entries for this property (3 total)
Method: DFT-HSE06 (alpha=0.25, omega=(0.11 Bohr radii)^-1)+ SOC
Origin: computational
Crystal system: triclinic
| Band gap (fundamental), eV |
|---|
Sample type: single crystal
- data set 225 (atomic structure)
- data set 745 (atomic structure)
- data set 1885 (atomic structure)
- data set 1901 (atomic structure)
- data set 1903 (atomic structure)
- data set 2269 (band structure)
Code: FHI-aims
Level of theory: Spin-orbit coupled hybrid DFT
Exchange-correlation functional: HSE06 functional; exchange mixing parameter: 0.25, screening parameter: 0.11 (Bohr radii)^(-1)
K-point grid: 3x7x7
Level of relativity: Spin-orbit coupling included as follows: Self-consistent scalar relativity (atomic zero-order regular approximation) with spin-orbit coupling applied non-selfconsistently in the energy band structure calculation.
Basis set definition: All-electron; "intermediate" numerical settings and basis sets.
Numerical accuracy: Note that DFT-computed energy band gap values, even at the level of DFT-HS06+SOC, are not intended to capture the experimentally correct fundamental gap with quantitative accuracy. Rather, they are collected be comparable to other computational band gaps at the same level of theory in order to capture trends between different sources.
External repositories:
Comment: The geometry used was computationally optimized (unit cell and atomic positions) starting from the XRD-determined structure reported in https://doi.org/10.1021/acs.inorgchem.7b01094 . The level of theory used was DFT-PBE including the Tkatchenko-Scheffler van der Waals correction. The structure is available in the HybriD3 database as dataset number 745.
Entry added on: March 6, 2023, 11:45 a.m.
Entry added by: Volker Blum Duke University
Last updated on: March 6, 2023, 11:56 a.m.
Last updated by: Volker Blum Duke University
Download data
Band structure
Method: DFT-HSE06 (alpha=0.25, omega=(0.11 Bohr radii)^-1)+ SOC
Origin: computational
Crystal system: triclinic
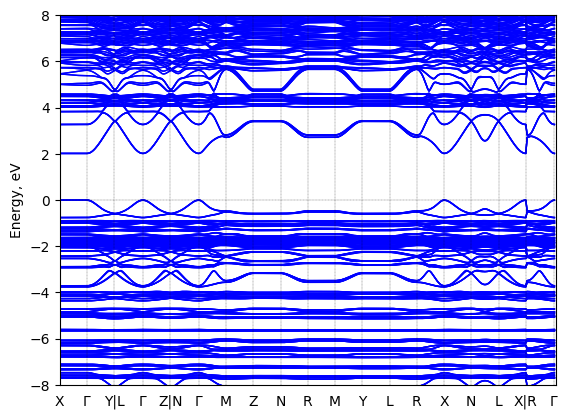
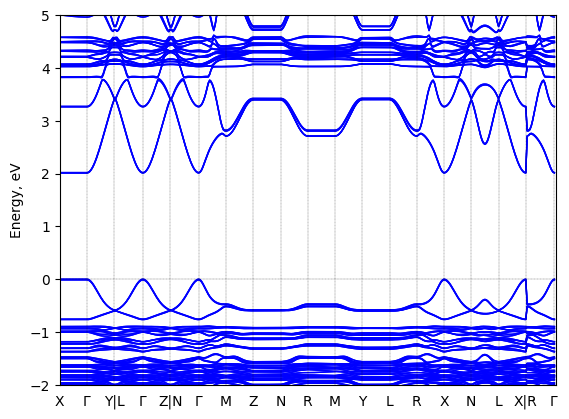
Sample type: single crystal
- data set 225 (atomic structure)
- data set 745 (atomic structure)
- data set 1885 (atomic structure)
- data set 1901 (atomic structure)
- data set 1903 (atomic structure)
- data set 2267 (band gap (fundamental))
Code: FHI-aims
Level of theory: Spin-orbit coupled hybrid DFT
Exchange-correlation functional: HSE06 functional; exchange mixing parameter: 0.25, screening parameter: 0.11 (Bohr radii)^(-1)
K-point grid: 3x7x7
Level of relativity: Spin-orbit coupling included as follows: Self-consistent scalar relativity (atomic zero-order regular approximation) with spin-orbit coupling applied non-selfconsistently in the energy band structure calculation.
Basis set definition: All-electron; "intermediate" numerical settings and basis sets.
Numerical accuracy: Note that DFT-computed energy band gap values, even at the level of DFT-HS06+SOC, are not intended to capture the experimentally correct fundamental gap with quantitative accuracy. Rather, they are collected be comparable to other computational band gaps at the same level of theory in order to capture trends between different sources.
Geometry used in the calculation
Comment: The geometry used was computationally optimized (unit cell and atomic positions) starting from the XRD-determined structure reported in https://doi.org/10.1021/acs.inorgchem.7b01094 . The level of theory used was DFT-PBE including the Tkatchenko-Scheffler van der Waals correction. The structure is available in the HybriD3 database as dataset number 745.
Entry added on: March 7, 2023, 11:32 a.m.
Entry added by: Xixi Qin Duke University
Last updated on: March 7, 2023, 11:35 a.m.
Last updated by: Xixi Qin Duke University
Download data
Atomic structure
See all entries for this property (31 total)
Method: Single-crystal X-ray diffraction
Origin: experimental (T = 298.0 K)
Space group: P -1
Crystal system: triclinic
| a: | 8.7389 (±0.0002) Å |
| b: | 8.7403 (±0.0002) Å |
| c: | 32.9952 (±0.0006) Å |
| α: | 84.646 (±0.001)° |
| β: | 84.657 (±0.001)° |
| γ: | 89.643 (±0.001)° |
- temperature = 298.0 K
Sample type: single crystal
- data set 745 (atomic structure)
- data set 1885 (atomic structure)
- data set 1901 (atomic structure)
- data set 1903 (atomic structure)
- data set 2267 (band gap (fundamental))
- data set 2269 (band structure)
Starting materials: PbI2 (99.999% trace metal basis), HI (57 wt % in H2O, with hypophosphorous acid as stabilizer, assay 99.95%), CH3OH (>99.9%), 2-phenylethylamine (PEA, 99%)
Product: Red and laminar crystals.
Description: Dissolve PbI2 (54.6 mg) in 0.5 mL of HI (57%). Place CH3OH (1 ml) on the top of the PbI2 solution. Add 0.030 mL of PEA liquid into the CH3OH layer. Crystals would form in the solution overnight.
Method: Single-crystal X-ray diffraction
Description: Single-crystal X-ray diffraction data were collected using a Bruker D8 ADVANCE Series II at room temperature. The crystal structures were solved and refined by Shelxl and Olex software.
Comment: This is the experimentally resolved structure, which contains a statistical (disordered) representation of the equatorial iodine atoms in the original refinement. This structure is our recommended "best" published experimental structure for phenethylammonium lead iodide at room temperature, supported by a computational comparison of the total energies of various published structures after full structure optimization in Chemistry of Materials, Vol. 34, issue 7, 3109-3122 (2022), https://doi.org/10.1021/acs.chemmater.1c04213. The computationally relaxed (PEA)2PbI4 structure with resolved disorder (i.e., no overlapping iodine positions), optimized using the PBE+Tkatchenko-Scheffler approach, is also available in the HybriD3 database (see datasets linked to the present one.)
Entry added on: April 15, 2019, 9:54 p.m.
Entry added by: Xiaochen Du Duke University
Last updated on: Sept. 10, 2024, 4:09 p.m.
Last updated by: Volker Blum Duke University
Download data